For our international customers, please be advised that orders cannot be placed through our website by customers in countries with International Distributor representation.
Chymotrypsinogen A - Manual
Source:
Bovine Pancreas
CAS:
9035-75-0
{kind=link}
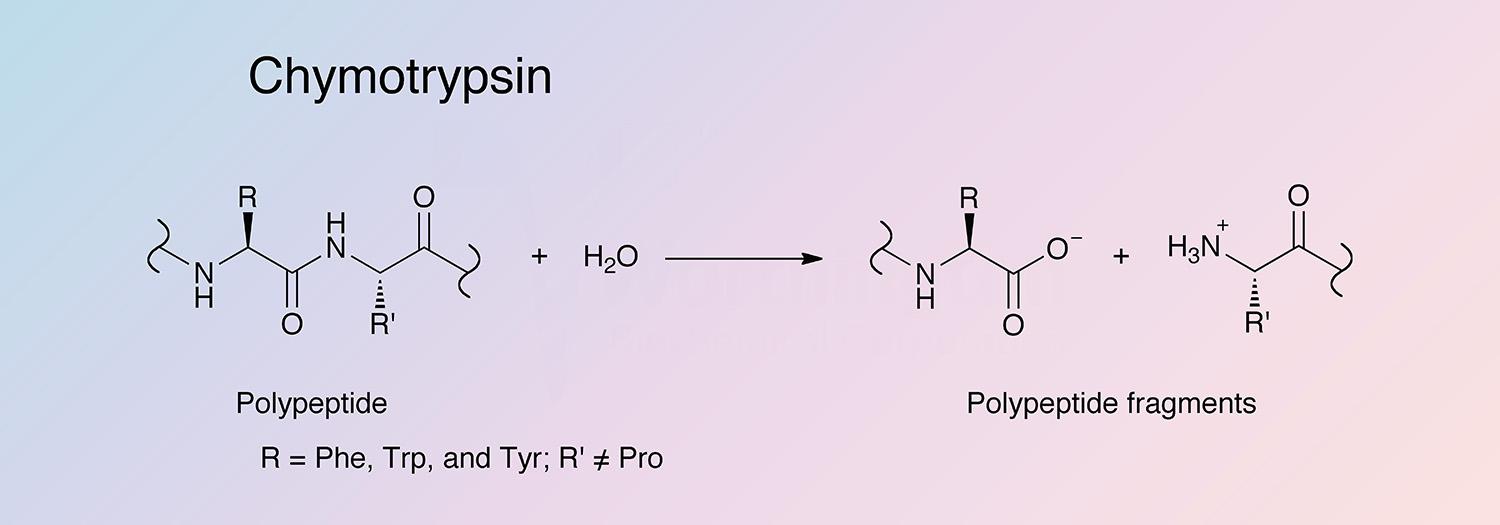
Chymotrypsin is a serine endopeptidase produced by the acinar cells of the pancreas. Chymotrypsin becomes activated after proteolysis of chymotrypsinogen by trypsin. While trypsin hydrolyzes at lysine and arginine, chymotrypsin selectively cleaves peptide bonds formed by aromatic residues (tyrosine, phenylalanine, and tryptophan) (Hedstrom et al. 1992). Two predominant forms of chymotrypsin, A and B, are found in equal amounts in cattle pancreas. They are very similar proteins (80% identical), but have significantly different proteolytic characteristics (Hartley 1964, Meloun et al. 1966, Smillie et al. 1968, and Gráf et al. 2004). The information below pertains primarily to the A form of chymotrypsinogen and chymotrypsin.
History
In the early 1900s, Vernon proposed that pancreatic preparations could give rise to an intrinsic activator of its own enzymes (Vernon 1901). Vernon’s milk-clotting experiments determined there were at least two enzymes present and that one was more stable than the other (Vernon 1902). However, this idea was not widely accepted until 1934 when Kunitz and Northrop confirmed the presence of an enzyme in addition to trypsin, naming it chymotrypsin. They were able to crystallize chymotrypsin, as well as the inactive precursor, chymotrypsinogen (Kunitz and Northrop 1934). In 1938, Kunitz isolated different active forms of chymotrypsin, designating them as alpha, beta, and gamma (Kunitz 1938).
In the early 1940s Fruton and Bergmann further studied the specificity of chymotrypsin, reporting on several new substrates (Fruton and Bergmann 1942). Jacobsen soon identified additional forms of chymotrypsin, designating them as delta and pi (Jacobsen 1947). In 1948, Schwert further characterized the molecular weights of chymotrypsin and chymotrypsinogen.
In 1954, the first evidence for the three-step mechanism of chymotrypsin hydrolyzing amide and ester substrates was reported on by Hartley and Kilby, who hypothesized the presence of an acyl enzyme intermediate, which was later proven to be true (Henderson 1970). In 1955, Laskowski obtained a second crystalline chymotrypsinogen, naming it chymotrypsinogen B. In 1964 Hartley determined the amino acid sequence of chymotrypsin A, which was later refined by Meloun et al. in 1966. In 1968, Smillie et al. determined the amino acid sequence of chymotrypsin B, which revealed 80% sequence identity with chymotrypsin A. Throughout the 1970s and 1980s research was done to better understand the mechanism of action, and identify the differences in amino acid sequences between trypsin and chymotrypsin (Steitz et al. 1969, Cohen et al.1981, Asbóth and Polgár 1983, and Gráf et al. 1988).
In the 1990s, chymotrypsin was purified from other sources including Atlantic cod (Ásgeirsson and Bjarnason 1991), and camel (Al-Ajlan and Bailey 1997). Work also begun on investigating inhibitors (Baek et al. 1990), and Frigerio et al. elucidated the crystal structure of bovine chymotrypsin to a 2.0 Å resolution (Frigerio et al. 1992).
Recent research has investigated the folding and denaturation of chymotrypsin over a range of concentrations (Ghaouar et al. 2010), chymotrypsin’s interaction with nanoparticle substrates (You et al. 2006, and Jordan et al. 2009), and increasing chymotrypsin stability by conjugating to PEG molecules (Castellanos et al. 2005, and Rodríguez-Martínez et al. 2009).
Specificity
Chymotrypsin is activated through cleavage of the bond between arginine and isoleucine (R15 and I16) by trypsin, causing structural modifications and formation of the substrate binding site (Sears 2010). Chymotrypsin differs from trypsin in that trypsin cleaves peptides at arginine and lysine residues, while chymotrypsin prefers large hydrophobic residues (Hedstrom et al. 1992). Chymotrypsin preferentially catalyzes the hydrolysis of peptide bonds involving L-isomers of tyrosine, phenylalanine, and tryptophan. It also readily acts upon amides and esters of susceptible amino acids. Chymotrypsin’s specificity for large hydrophobic residues can be explained by a hydrophobic S1 binding pocked formed by residues 189 through 195, 214 through 220, and 225 through 228 (Cohen et al. 1981).
Although the structure of trypsin and chymotrypsin’s S1 site show only one difference (at position 189), site-directed mutagenesis of trypsin and chymotrypsin have failed to interchange specificities, suggesting the mechanism by which trypsin and chymotrypsin achieve substrate specific catalysis is not fully understood (Steitz et al. 1969, and Gráf et al. 1988).
Composition
The three amino acid residues of the catalytic triad (H57, D102, and S195) are essential for peptide bond cleavage and are stabilized by hydrogen bonds (Sears 2010, and Gráf et al. 2004). G193 and S195 make up the oxyanion hole and interact with the carbonyl group of the scissile peptide bond, orienting it to form the tetrahedral intermediate (Rühlmann et al. 1973, Huber and Bode 1978, and Gráf et al. 2004).
Molecular Characteristics
Chymotrypsin A and B share 80% sequence identity (Hartley 1964, Meloun et al. 1966, Smillie et al. 1968, and Gráf et al. 2004). The amino acids of the catalytic triad (H57, D102, and S195) are highly conserved in the sequences of the peptidases of family S1 (Gráf et al. 2004). The serine at position 214 is also highly conserved in the family and has been proposed as the fourth member of the catalytic triad (Ohara et al. 1989, and McGrath et al. 1992).
Applications
- Sequence analysis
- Peptide synthesis
- Peptide mapping
- Peptide fingerprinting
Characteristics of Chymotrypsinogen A
Protein Accession Number
P00766
CATH Classification
- Class: Mainly Beta
- Architecture: Beta Barrel
- Topology: Trypsin-like Serine Protease
Molecular Weight
- 25.6 kDa (Wilcox 1970)
Optimal pH
7.8-8.0 (Rick 1974)
Isoelectric point
- 8.52 (Chymotrypsinogen, Theoretical)
- 8.33 (Chymotrypsin, Theoretical)
Extinction Coefficient
- 51,840 cm-1 M-1 (Theoretical)
- E1%,280 = 20.19 (Chymotrypsinogen, Theoretical)
- E1%,280 = 20.57 (Chymotrypsin, Theoretical)
Active Site Residues
- Histidine (H57)
- Aspartate (D102)
- Serine (S195)
Activators
- Cetyltributylammonium bromide (Spreti et al. 2008)
- Dodecyltrimethylammonium bromide (Abuin et al. 2005)
- Hexadecyltrimethylammonium bromide (Celej et al. 2004)
- Tetrabutylammonium bromide (Spreti et al. 2001)
Inhibitors
- Hydroxymethylpyrroles (Abell and Nabbs 2001)
- Boronic acids (Smoum et al. 2003)
- Courmarin derivatives (Pochet et al. 2000)
- Peptidyl aldehydes (Lesner et al. 2009)
- Peptides from natural sources (Telang et al. 2009, Roussel et al. 2001, and Chopin et al. 2000)
- Peptides containing an unnatural amino acid (Legowska et al. 2009, and Wysocka et al. 2008)
Chymotrypsinogen A Products
Description
Activity
Code
Cat. #
Size
Price
Description
Chymotrypsinogen A, Purified
Source:
Bovine Pancreas
Five times crystallized, electrophoretically homogeneous. Supplied as a dialyzed, lyophilized powder.
Intrinsic activity <0.55% chymotrypsin.
Intrinsic activity <0.55% chymotrypsin.
Store at 2-8°C.
Code
CGC
Product details
LS005630
1 gm
$74.00
LS005623
5 gm
$260.00
LS005622
Bulk
---