For our international customers, please be advised that orders cannot be placed through our website by customers in countries with International Distributor representation.
Ribonuclease A - Manual
Source:
Bovine Pancreas
CAS:
9001-99-4
EC:
3.1.27.5
{kind=link}
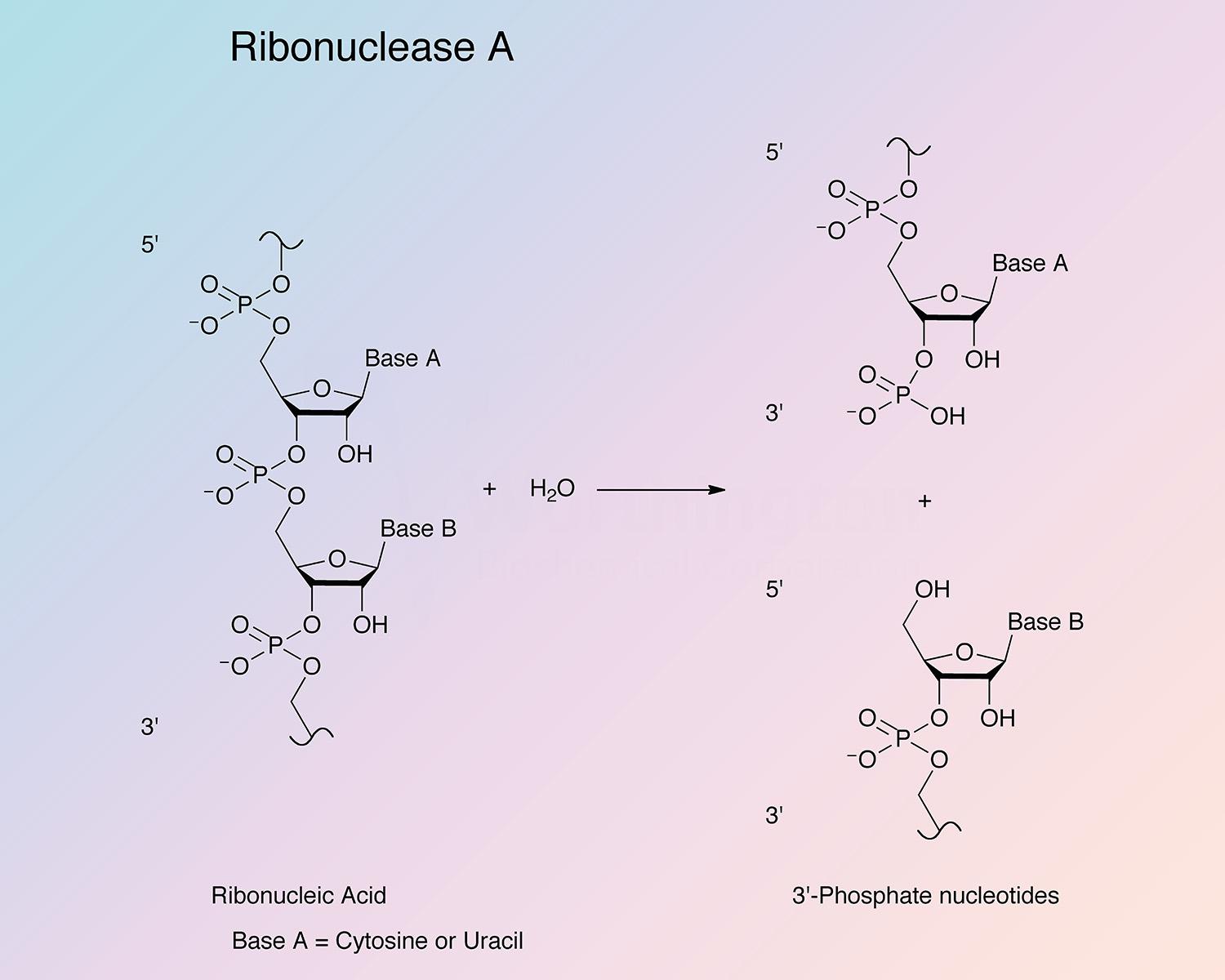
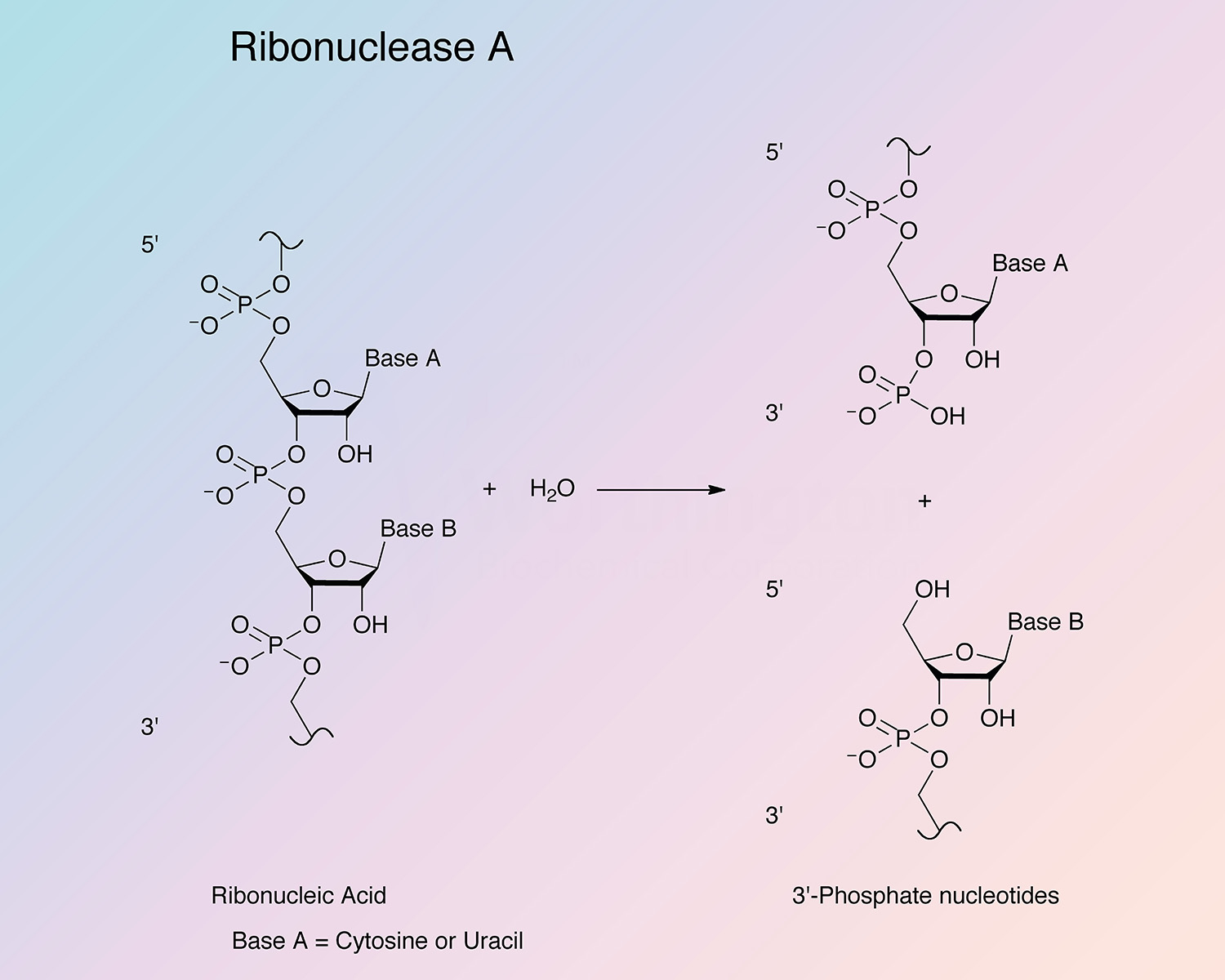
Pancreatic ribonuclease (RNase) is an endoribonuclease. It catalyzes the cleavage of the phosphodiester bond between the 5’-ribose of a nucleotide and the phosphate group attached to the 3’-ribose of an adjacent pyrimidine nucleotide. This cleavage forms a 2’,3’-cyclic phosphate, which is then hydrolyzed to the corresponding 3’-nucleoside phosphate.
RNase is found in greatest quantity in ruminant pancrease (Barnard 1969). The major component of the crystalline enzyme is RNase A; a minor component is RNase B. RNase B is the glycosylated form of RNase A (Beintema et al. 1976).
History
The work of Jones in 1920 is usually cited as the “beginning” of pancreatic ribonuclease (Richards and Wycoff 1971). RNase was isolated by Dubos and Thompson in 1938 and crystallized by Kunitz in 1940.
In 1947 Worthington was the first company to manufacture highly purified crystalline RNase. In the early 1950s, the company Armour prepared crude crystalline enzyme, and offered it at a very affordable price. Through the 1960s and 1970s, RNase A was a favorite to study primarly because it is remarkably thermostable and present at high concentration in an accessible source, bovine pancreas. These studies led to the elucidation of the crystal structure (Anfinsen 1959, Groves 1966, and Scheraga 1967), determination of the amino acid sequence (Smyth et al. 1963), identification of the catalytic mechanism (Beers 1960), and clarification of folding pathways (Hantgan et al. 1974). RNase A was the first enzyme and third protein for which a correct amino acid sequence was determined (Raines 1998).
Four Nobel prizes have been awarded for work associated with studies of RNase (Anfinsen, Moore, Stein, and Merrifield). The vast literature and numerous studies have made RNase the most extensively studied enzyme of the 20th century (Raines 1998).
Recent work continues to investigate the synthesis and maturation of RNase in the endoplasmic reticulum of live cells (Geiger et al. 2010). Much work is also still being dedicated to studying the folding and aggregation of RNase (Benito et al. 2008, Iwaoka et al. 2008, and Arai et al. 2010). The enzyme’s role in cancer development and gene regulation is being studied (Shlyakhovenko 2009), and it is being developed into cancer chemotherapeutic agents (Chao et al. 2010).
Specificity
RNase A is specific for pyrimidine nucleoside linkages (Volkin and Cohn 1953). The reaction is believed to take place in two steps. In the first step, the 3’,5’-phosphodiester bond is cleaved, while generating a 2’,3’-cyclic phosphodiester intermediate. In the second step, the cyclic phosphodiester is hydrolyzed to a 3’-monophosphate group. The first step is nonspecific with respect to the nitrogenous base of the substrate; however, the second step is absolutely specific for pyrimidine nucleotides with terminal 2’,3’-cyclic phosphates. RNase B has the same specificity as RNase A toward both cyclic cytidylate and yeast RNA (Plummer and Hirs 1963). RNase A shows a preference for larger substrates (Nogués et al. 1995).
The enzyme cleaves at cytidine residues twice as fast as at uridyl residues (Richards and Wyckoff 1971). Thr45 has been found to be most important for mediating the pyrimidine specificity, both by forming hydrogen bonds with pyrimidine bases and sterically excluding purine bases (del Cardayré and Raines 1994). The side chain of Asp83 is important for stabilizing the transition state during the cleavage of uridine-containing substrates; this residue has no effect on the kinetics of cytidine cleavage (del Cardayré and Raines 1995).
Composition
The shape of the protein resembles a kidney, with the active site residues laying in the cleft (Richardson 1981, and Raines 1998). The secondary structure contains long four-stranded anti-parallel beta-sheets and three short alpha-helices (Raines 1998). RNase A contains four disulfide bonds, which are critical to the stability of the native enzyme. Two of these disulfide bonds lie between an alpha-helix and a beta-sheet and contribute more to the thermal stability than do the other two (Klink et al. 2000). RNase B is a glycoprotein containing at Asn34 a single oligosaccharide composed of six residues of mannose and two residues of N-acetylglucosamine (Tarentino et al. 1970).
Molecular Characteristics
RNase A is a small protein, the mature enzyme only having 124 amino acid residues, with no carbohydrate attached. RNase A contains 19 of the 20 amino acids, lacking only tryptophan (Nogués et al. 1995, and Raines 1998). The three dimensional structure of RNase A is fully encoded by its amino acid sequence (White and Anfinsen 1959, and Raines 1998). All eight human RNase A-like genes are located on chromosome 14. Each encodes a secretory signal sequence and contains an invariant catalytic triad of two histidines and one lysine with a conserved motif (CKXXNTF) (Marshall et al. 2008).
The amino acid sequences of many RNase A homologues have been identified, making RNase A a model system for vertebrate molecular evolution (Dyer and Rosenberg 2006). From the sequences and their distribution over a range of species it has been established that RNase A is a modern protein that is evolving rapidly (Doolittle 1992, and Raines 1998).
Applications
- RNA removal during DNA isolation
- RNA sequence analysis
- RNase protection assays
- RNA quantification or mapping
- Purifying plasmid DNA
- Genomic DNA isolation
- Molecular weight marker
Characteristics of Ribonuclease A
Protein Accession Number
P61823
CATH Classification
- Class: Alpha-Beta
- Architecture: Roll
- Topology: P-30 Protein
Molecular Weight
- RNase A: 13.7 kDa (Hirs et al. 1956b)
- RNase B: 14.700 ± 0.3 (Plummer and Hirs 1963)
Optimal pH
RNase A: 7.0-7.5 (Brown and Todd 1955)
Isoelectric point
- RNase A: 9.3 (Ui 1971)
Extinction Coefficient
- RNase A and B: 8,640 cm-1M-1 (Theoretical)
- RNase A: E 1%, 280 = 7.3 (Worthington RNase A)
- RNase B: E 1%, 280 = 5.77 (Theoretical, RNase B)
Active Site Residues
- Histidine (H12, H119)
- Lysine (K41)
Activators
- Sodium chloride (Weickmann et al. 1981)
- Sulfate (Moosavi-Movahedi et al. 2006)
Inhibitors:
- Heavy metal ions
- Ribonuclease inhibitor (RI), a 50 kDa protein that constitutes ≤ 0.01% of the protein in the cytosol of mammalian cells (Takahashi 1967)
- Uridine-vanadate complexes (Lindquist et al. 1973)
Ribonuclease A Products
Description
Activity
Code
Cat. #
Size
Price
Description
Ribonuclease A, DNase & Protease Free
Source:
Bovine Pancreas
Molecular Biology Grade. A solution containing approximately 5 mg/ml in 50% glycerol. Prepared specifically for use in purifying DNA plasmids. Each lot is assayed for DNase and protease.
Store at 2-8°C or -20°C.
Code
RPDF
Product details
LS002131
1 mg
$28.00
LS002132
5 mg
$92.00
LS002130
Bulk
---
Description
Ribonuclease A, Purified
Source:
Bovine Pancreas
A lyophilized preparation equivalent to Code: RASE. Contains some aggregates as a result of lyophilization but shows no difference in specific activity.
Store at 2-8°C. PROTECT FROM MOISTURE.
Code
RAF
Product details
LS005649
25 mg
$70.00
LS005650
100 mg
$225.00
LS005655
Bulk
---
Description
Ribonuclease A, Purified Solution
Source:
Bovine Pancreas
Chromatographically purified. Aggregate-free. (Monophoretic on native gel electrophoresis.) A liquid in 100mM phosphate buffer containing 0.1% v/v phenol as a preservative. Note: The presence of 100mM phosphate makes this preparation unsuitable for heat treatment.
Store at -20°C.
Dry Ice required
Code
RASE
Product details
LS005677
25 mg
$64.00
LS005679
100 mg
$164.00
LS005681
Bulk
---
Description
Ribonuclease A
Source:
Bovine Pancreas
A chromatographically purified, lyophilized powder.
Store at 2-8°C. PROTECT FROM MOISTURE.
Code
R
Product details
LS003431
200 mg
$112.00
LS003433
1 gm
$480.00
LS003435
Bulk
---
Description
Ribonuclease B
Source:
Bovine Pancreas
A partially purified preparation containing a mixture of RNase A and RNase B. A soluble, dialyzed lyophilized powder.
Store at 2-8°C.
Code
RB
Product details
LS005710
100 mg
$118.00
LS005715
Bulk
---
Description
Ribonuclease A, Recombinant, Protease & DNase Free, AF
Source:
Komagataella phaffii (previously P. pastoris)
Recombinant bovine pancreatic Ribonuclease A produced in Komagataella phaffii. Chromatographically purified, free of animal derived components, DNases and Proteases. Supplied as lyophilized powder.
Store at 2-8°C. PROTECT FROM MOISTURE.
Code
RRA1
Product details
LS01506
10 ku
$175.00
LS01508
25 ku
$350.00
LS01510
Bulk
---
Description
Ribonuclease A, Recombinant, AF
Source:
Komagataella phaffii (previously P. pastoris)
Recombinant bovine pancreatic Ribonuclease A produced in Komagataella phaffii. For the removal of RNA in bioprocessing applications. May contain DNases and Proteases. Supplied as a lyophilized powder, Animal Free/AF.
Store at 2-8°C. PROTECT FROM MOISTURE.
Code
RRA2
Product details
Minimum order quantity
20000
LS01516
Bulk
---
LS01512
100 mg
$115.00
LS01514
1000 mg
$525.00